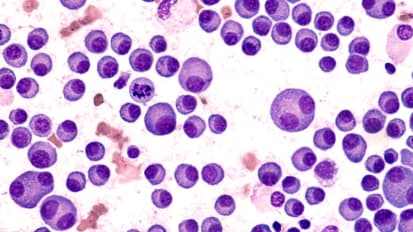
Finding New Weapons: Novel Treatment Approaches in Myeloma
Kelvin Lee, MD, gives a “philosophical talk” regarding novel treatment approaches in myeloma. This lecture helps illustrate the way doctors think about drugs and treatments for treating myeloma.